1. Herrick JB. Peculiar Elongated and Sickle-shaped Red Blood Corpuscles in a Case of Severe Anemia. Yale J Biol Med 2001;74:179-184.
2. Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 1995;332(20):1317-22. (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7715639
http://www.nejm.org/doi/pdf/10.1056/NEJM199505183322001).
3. Ferster A, Vermylen C, Cornu G, et al. Hydroxyurea for treatment of severe sickle cell anemia: a pediatric clinical trial. Blood 1996;88(6):1960-4. (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=8822914
http://bloodjournal.hematologylibrary.org/content/88/6/1960.full.pdf).
4. Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 2011;377(9778):1663-72. DOI: 10.1016/S0140-6736(11)60355-3.
5. de Montalembert M, Voskaridou E, Oevermann L, et al. Real-Life experience with hydroxyurea in patients with sickle cell disease: Results from the prospective ESCORT-HU cohort study. Am J Hematol 2021;96(10):1223-1231. DOI: 10.1002/ajh.26286.
6. Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol 2010;85(6):403-8. (In eng). DOI: 10.1002/ajh.21699.
7. Voskaridou E, Christoulas D, Bilalis A, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (LaSHS). Blood 2010;115(12):2354-63. (In eng). DOI: 10.1182/blood-2009-05-221333.
8. Lobo CL, Pinto JF, Nascimento EM, Moura PG, Cardoso GP, Hankins JS. The effect of hydroxcarbamide therapy on survival of children with sickle cell disease. Br J Haematol 2013;161(6):852-60. (Clinical Trial
Research Support, Non-U.S. Gov’t). DOI: 10.1111/bjh.12323.
9. Buchanan G, Yawn BP. Evidence Based Management of Sickle Cell Disease. NIH Consens State Sci Statements 2014.
10. Ware RE, Schultz WH, Yovetich N, et al. Stroke With Transfusions Changing to Hydroxyurea (SWiTCH): a phase III randomized clinical trial for treatment of children with sickle cell anemia, stroke, and iron overload. Pediatr Blood Cancer 2011;57(6):1011-7. (In eng). DOI: 10.1002/pbc.23145.
11. McGann PT, Ware RE. Hydroxyurea therapy for sickle cell anemia. Expert Opin Drug Saf 2015;14(11):1749-58. DOI: 10.1517/14740338.2015.1088827.
12. DeBaun MR. Hydroxyurea therapy contributes to infertility in adult men with sickle cell disease: a review. Expert Rev Hematol 2014;7(6):767-73. DOI: 10.1586/17474086.2014.959922.
13. Joseph L, Jean C, Manceau S, et al. Effect of hydroxyurea exposure before puberty on sperm parameters in males with sickle cell disease. Blood 2021;137(6):826-829. DOI: 10.1182/blood.2020006270.
14. Pecker LH, Hussain S, Christianson MS, Lanzkron S. Hydroxycarbamide exposure and ovarian reserve in women with sickle cell disease in the Multicenter Study of Hydroxycarbamide. Br J Haematol 2020;191(5):880-887. DOI: 10.1111/bjh.16976.
15. McGann PT, Ware RE. Hydroxyurea for sickle cell anemia: what have we learned and what questions still remain? Curr Opin Hematol 2011;18(3):158-65. DOI: 10.1097/MOH.0b013e32834521dd.
16. Lederman HM, Connolly MA, Kalpatthi R, et al. Immunologic effects of hydroxyurea in sickle cell anemia. Pediatrics 2014;134(4):686-95. DOI: 10.1542/peds.2014-0571.
17. Adekile AD, Gupta R, Al-Khayat A, Mohammed A, Atyani S, Thomas D. Risk of avascular necrosis of the femoral head in children with sickle cell disease on hydroxyurea: MRI evaluation. Pediatr Blood Cancer 2019;66(2):e27503. DOI: 10.1002/pbc.27503.
18. Luchtman-Jones L, Pressel S, Hilliard L, et al. Effects of hydroxyurea treatment for patients with hemoglobin SC disease. Am J Hematol 2016;91(2):238-42. DOI: 10.1002/ajh.24255.

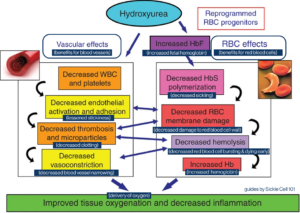 Hydroxyurea use was first studied and reported in adults who had sickle cell anemia (SS or S beta zero thalassemia), it was found to decrease the number of hospitalizations for pain crises and decrease the frequency of acute chest syndrome by half, and also decrease transfusion requirements2.
Hydroxyurea use was first studied and reported in adults who had sickle cell anemia (SS or S beta zero thalassemia), it was found to decrease the number of hospitalizations for pain crises and decrease the frequency of acute chest syndrome by half, and also decrease transfusion requirements2.