Welcome to Our Learn Center
Learn all about sickle cell disease and sickle cell trait through videos, frequently asked questions, Sicklepedia, fact sheets, resources and more.
 Videos
Videos FAQ
FAQ Sicklepedia
Sicklepedia Fact Sheets
Fact Sheets
Healthy Living with Sickle Cell
Fellow warrior and Sickle Cell 101 executive director, Cass Trimnell shares healthy living tips to better manage sickle cell disease.
Curative and Emerging Therapies for Sickle Cell Disease
Med student and SC101 team member Abi Oshiobugie Suleiman discusses curative options for sickle cell diseased and treatments that are in the horizon.
Treatments for Sickle Cell Disease
Fellow warrior and Sickle Cell 101 executive director, Cass Trimnell shares therapies that slows the progression of sickle cell disease.
Treating Sickle Cell Symptoms
Fellow warrior and Dr. Terry Jackson, PhD shares options for treating sickle cell symptoms.
FAQ
Avascular necrosis is a common problem for people who have sickle cell disease.
An orthopedic physician can assess damage caused by AVN, a physiatrist who can guide a physical therapy program that could help with mobility and decrease pain, and a physical therapist can guide you through those exercises
Often times, people opt for a core decompression procedure, and if that does not work a hip replacement surgery.
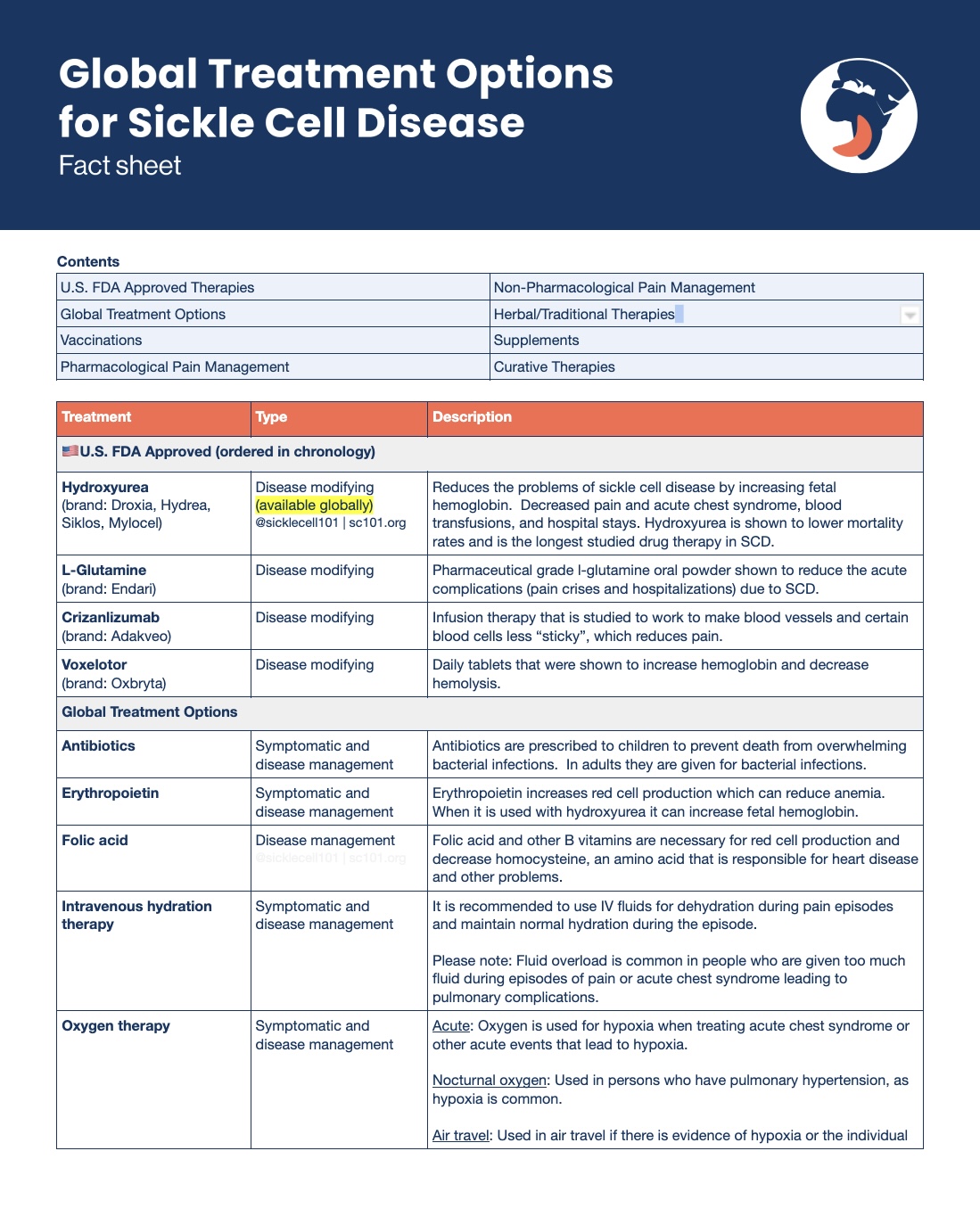
Medications for sickle cell disease can be categorized into the following groups:
- • Preventative therapies to keep you healthy (i.e. antibiotics, folic acid)
- • Pain medications to treat acute and chronic pain
- • Disease modifying therapies, which tackle the underlying causes of the disorder
There are currently four U.S. FDA approved drug therapies specifically for sickle cell disease. They include:
- 1.) Hydroxyurea
- 2.) Pharmaceutical grade l-glutamine
- 3.) Crizanlizumab
- 4.) Voxelotor
Hydroxyurea has been used medically in the United States since 1967 and was approved for use in sickle cell disease in 1998.
Hydroxyurea was chosen to be studied in sickle cell disease due to the fact that it is an oral medication and has been used for decades and has mild side effects that are reversible when the medication is stopped.
Hydroxyurea is known to be a safe and low-risk medication for sickle cell disease. When considering new therapies, always look at the risk-benefit of medications and treatments in medicine. This is a decision to be made with your healthcare provider. It is important to remember hydroxyurea must be monitored on a regular basis, your healthcare provider must be familiar with the use of this medication.
Stem cell transplant/bone marrow transplant is a curative treatment option for sickle cell disease. Another emerging curative approach is gene therapy.
There are studies looking at transplant and gene therapy for adults with sickle cell disease.
What is best for you? Only you and your primary care provider can sort that out. Everything in medical practice has positive and negative effects, benefit and risk. You can decide how much risk you are willing to accept with a transplant.
When you are looking at studies see who is running the study. Look them up online. You can ask what their track record is, how many transplants have they done for people with sickle cell disease? What were the outcomes? How many people have completed the study, have there been problems with outcome. If a study cannot give you any information about outcomes you have to weigh that in your decision.
The white part of the eye (sclera) can turn yellow if you have high levels of bilirubin. Bilirubin is a breakdown product of hemoglobin in red blood cells. The most common reason for having yellow eyes (doctors call this scleral icterus) is due to rapid blood breakdown in your body called hemolysis. Some people tend to have more hemolysis, some less.
People with more blood breakdown can have yellow eyes most of the time, or just when the blood breakdown is increased.
If you do not drink enough water that would not be the cause of your yellow eyes, but over time drinking water might decrease the yellow tint to your sclera. If this is new or more intense or you have other symptoms (tired, dark urine, pain), you need to see your health care provider as soon as possible.

Sicklepedia
An online patient-friendly encyclopedia
Sickle cell disorders are a group of inherited blood disorders that are caused by a mutation, or change in DNA. This change alters the way red blood cells function in individuals with the disorder. Sickle cell disorders are genetic, meaning they are passed down from parents to their children.
Normally, red blood cells are round and flexible and can travel easily through blood vessels. However, in those with sickle cell disease, each time red blood cells release oxygen to tissues and organs, they transform into fragile, sticky, crescent (or sickle) shaped red blood cells called sickle cells. This transformation weakens… (read more)

Sickle Cell Trait
An online patient-friendly encyclopediaSickle cell trait (SCT) is a condition in which a person inherits only one sickle cell gene – a typo (mutation) in the DNA for the gene responsible for carrying oxygen within red blood cells (hemoglobin – hb).
It is estimated that 1 in 12 African Americans have sickle cell trait. Over 300 million people have sickle cell trait worldwide. While sickle cell trait disproportionately affects individuals of black or African descent, people with sickle cell trait make up various races and ethnicities…. (read more)