Cause: Genetic changes
Specialty: Hematology
Most Common Symptoms: Pain, fatigue, anemia, dactylitis, fever, infections
Important Dates:
1910 First discovery in western medicine
1949 Sickle cell disease becomes known as a molecular disease
2006 The World Health Organization recognizes sickle cell disease as a global health problem
2008 The United Nations creates a resolution recognizing sickle cell disease as a public health problem and creates June 19 as World Sickle Cell Day
2009 The first celebrated World Sickle Cell Day on June 19, 2009
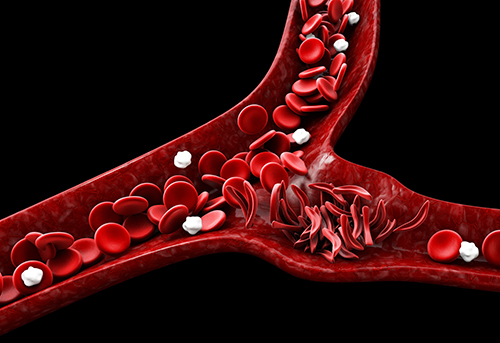
Sickle cell disorders are a group of inherited blood disorders that are caused by a mutation, or change in DNA. This change alters the way red blood cells function in individuals with the disorder. Sickle cell disorders are genetic, meaning they are passed down from parents to their children.
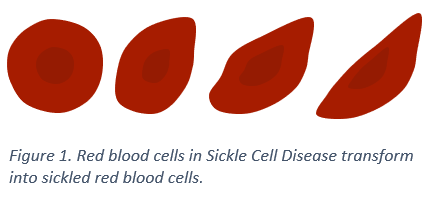
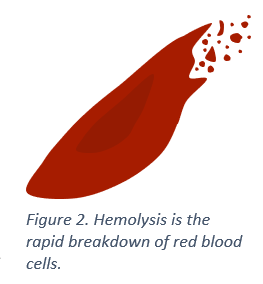
Normally, red blood cells are round and flexible and can travel easily through blood vessels. However, in those with sickle cell disease, each time red blood cells release oxygen to tissues and organs, they transform into fragile, sticky, crescent (or sickle) shaped red blood cells called sickle cells. This transformation weakens sickle cells over time and they break down at a faster rate (called hemolysis), which results in anemia and other complications.
The changes in shape and stickiness also make it difficult for sickle cells to move freely through small blood vessels. This can slow down blood flow or even cause blockage to the tissues they supply with oxygen. This event is called vaso-occlusion. The blockage and lack of oxygen cause tissue damage and inflammation which is the cause of painful vaso-occlusive events (pain crises) in sickle cell disease.
Blood flows through the entire body; therefore, any organ or tissue can be affected. When blood flow is reduced, oxygen is also reduced in the affected body parts and organs, which causes injury and even cell death. As a result, this causes damage to body parts that build up as time goes on.
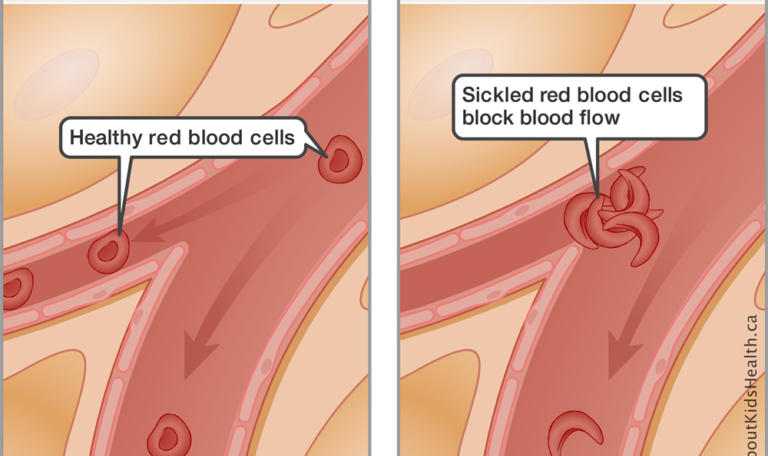
These events take place inside the body every second of every day, even if pain is not present. Outcomes of the disorder include extreme pain, damage to organs, and several other complications.