There are three main types of hemoglobin – embryonic, fetal, and adult.
- Embryonic hemoglobin, There are four embryonic hemoglobin molecules that are made during embryonic development. The last to form is fetal hemoglobin.
- Fetal hemoglobin is present in the fetus and in babies. Fetal hemoglobin binds to oxygen better than adult hemoglobin as before birth the baby obtains oxygen from their mother. After birth there is what is called a “hemoglobin switch” and adult hemoglobin is made and fetal hemoglobin declines.
- Adult hemoglobin usually replaces fetal hemoglobin at around the age of 6 months.
With the change to adult hemoglobin mutations in the alpha and beta globin molecules become apparent as these are the adult hemoglobin molecules. There are hundreds of hemoglobin mutations. Only a few cause disease. Some mutations lead to the absence of hemoglobin, these are the thalassemia mutations. Thalassemia mutations can be either in the beta or the alpha gene and can cause mild to severe disease depending on whether the gene is capable of making some hemoglobin or is unable to make any hemoglobin. In combination with hemoglobin S they cause sickle beta thalassemia. Alpha gene mutations can also occur in people who have sickle cell disease.
Other mutations change the hemoglobin itself these are the types of mutations that cause hemoglobin S and other types of hemoglobin that in combination with hemoglobin S that cause sickle cell disease. These structurally changed hemoglobin molecules are called “variants”. The common variant hemoglobin molecules are: hemoglobin C, hemoglobin D, and thalassemia mutations. There are other rare forms of sickle cell disease caused by other rare variants.
this is how sickle cell disease comes about. Sickle cell disease can be caused by multiple variants of hemoglobin, including Hemoglobin S, Hemoglobin C, Hemoglobin E, etc.
Hemoglobin S is the main hemoglobin variant in sickle cell disease. The mutation is in one of the beta subunits. The imbalance between alpha and beta subunits is called thalassemia. When the beta chain gene fails, it is called beta-thalassemia, because the type of thalassemia is determined by what molecule is abnormal. Mild thalassemia may present with few symptoms, while severe thalassemia can be life-threatening.

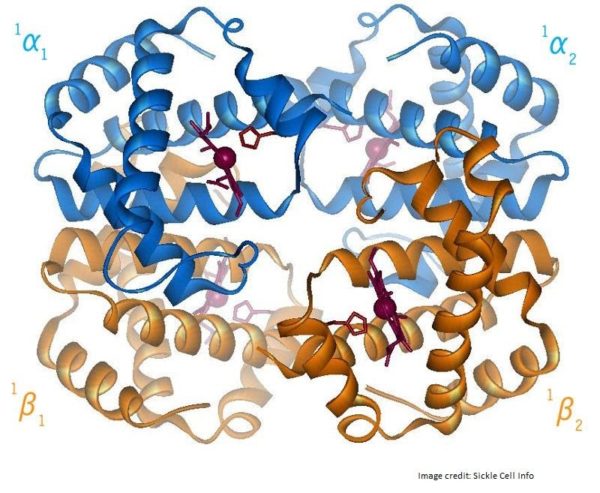
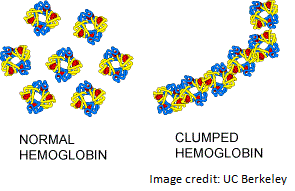 So, what causes these abnormal hemoglobin changes? Sickle cell is a genetic disease, meaning it is inherited from your parents. Hemoglobin is a protein and like all proteins it has a DNA code. This code is translated through RNA to amino acids that together form proteins within the cells. Sickle cell is a single mutation in the beta hemoglobin gene that changes this code for only one amino acid in the long chain that makes the beta globin protein. This single small change changes the properties of beta globin to allow polymerization leading to all of the problems with sickle cell disease.
So, what causes these abnormal hemoglobin changes? Sickle cell is a genetic disease, meaning it is inherited from your parents. Hemoglobin is a protein and like all proteins it has a DNA code. This code is translated through RNA to amino acids that together form proteins within the cells. Sickle cell is a single mutation in the beta hemoglobin gene that changes this code for only one amino acid in the long chain that makes the beta globin protein. This single small change changes the properties of beta globin to allow polymerization leading to all of the problems with sickle cell disease.