
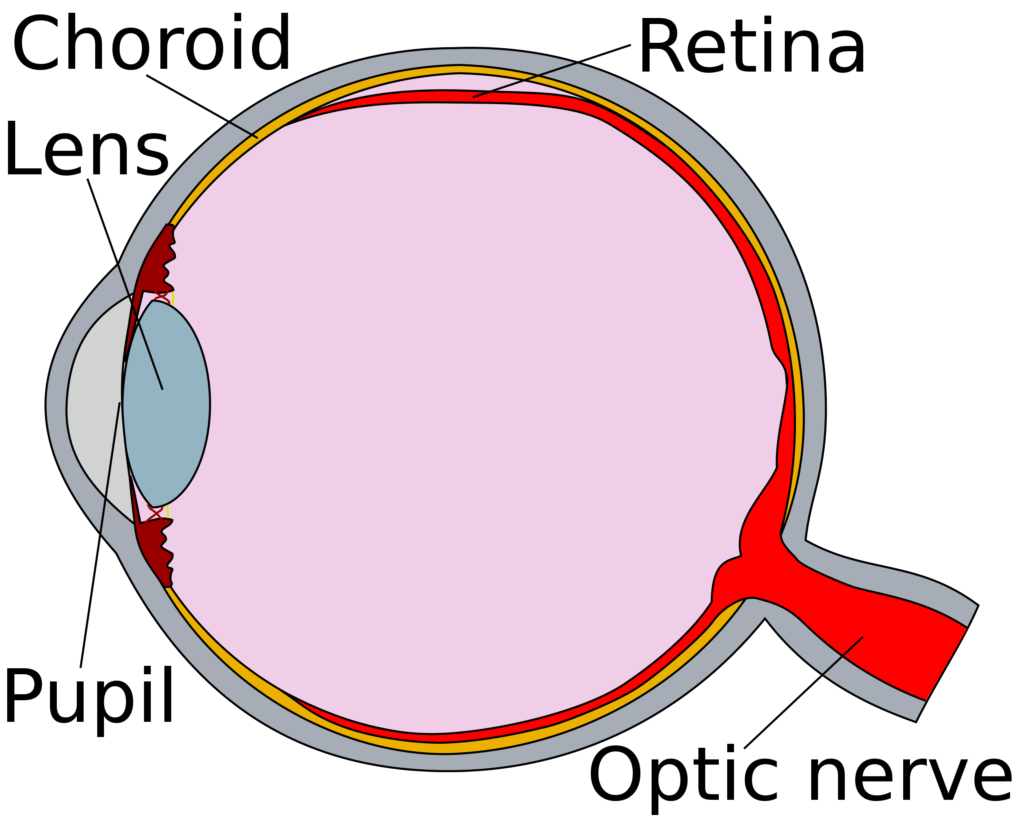
Sickle cell retinopathy is the separation of the retina and choroid within in eye, due to blockages in blood vessels caused by sickle red blood cells.
Retinopathy in sickle cell disease is caused by vessel occlusion and/or decreased blood flow due to increased viscosity – increased number of red blood cells – in the vessels of the retina. This vessel blockage leads to lowered levels of oxygen (hypoxia) and the increase of small vessels that are very unstable and not attached to the retina. These new vessels (neovascularization) are weaker and can rupture, causing bleeding into the eye.1 This can lead to the retina separating (detachment) and blindness.
These developments are painless and can go unnoticed by the patient unless bleeding (red blood cells) obscures the macula, which is where vision takes place. The signs of retinopathy are “floaters” which are red cells that can be seen in the visual field, areas of decreased vision seen as black spots or blurry vision.
Children rarely complain of these findings and retinopathy can begin at a young age. It is recommended that all children have a dilated eye examination starting at the age of 10 years and annually thereafter.
Over 40% of people who have hemoglobin SC will develop retinopathy in their lifetime and about 15% of people who have SS will develop retinopathy in their lifetime.