Hemolysis is the term given to when red blood cells break down. It’s an important underlying feature of sickle cell diseases.
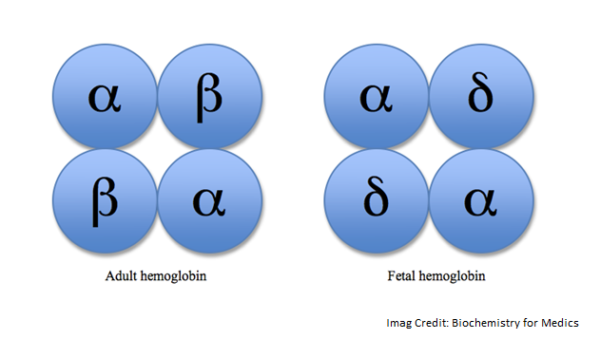
To understand why hemolysis happens in people with sickle cell disease, it’s useful to understand the basic structure of hemoglobin molecules.
Hemoglobin molecules have four subunits, two beta-globin, and two alpha-globin molecules. In normal hemoglobin, these four subunits are finely balanced to allow oxygen to bind and unbind without causing the hemoglobin to change shape.
Sickle cell disease causes a mutation (SS) in the two beta-globin subunits. In instances of low oxygen, the abnormal hemoglobin clumps to form long chains This is called “polymerization.” This in turn changes the structure of the red blood cell membrane. Unlike normal red blood cells, which are flexible and disc-shaped, sickle cells become rigid and sticky, sticking to each other and the blood vessel walls. When there is enough oxygen, sickle cells can resume their normal shape.
Have you ever broken a wire by bending it back and forth repeatedly? This is essentially what happens when sickle cells repeatedly change their shape. In time, they fracture and break apart. This breakdown is known as hemolysis. The normal lifespan of a red blood cell is 120 days, the lifespan of a sickle cell is just 14 days.
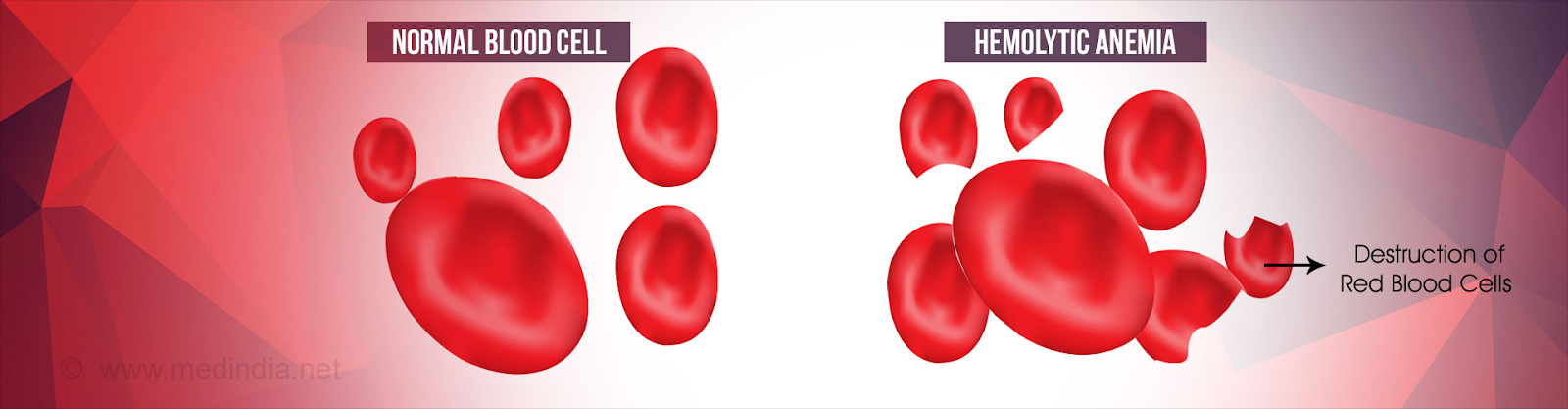
Hemolysis is considered an important part of the pathophysiology of sickle cell anemia. (The term pathophysiology relates to the processes within the body that contribute to a disease). The breakdown of red blood cells releases hemoglobin into the bloodstream that can lead to many complications. The most obvious result of hemolysis is anemia, due to the fact that the production of red blood cells can not keep up with the hemolysis.
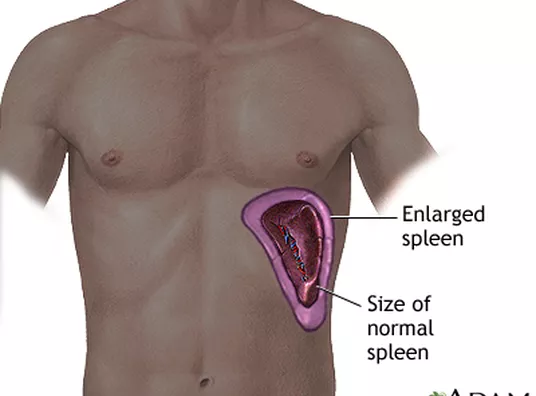 Splenic sequestration:
Splenic sequestration: